Reference: February 2024 | Issue 2 | Vol 10 | Page 14
Sarcomas are a heterogeneous group of rare malignancies arising from mesenchymal tissues such as bone, cartilage, muscle, and other connective tissues.1 They are much rarer than carcinoma, and tend to grow locally and invade adjacent tissues.
Sarcomas pose significant challenges in terms of diagnosis, classification, and treatment due to their diverse histology and variable clinical behaviour. Heterogeneity is highlighted by the identification of over 100 different sarcoma subtypes which vary in pathology, clinical presentation, molecular characteristics, and response to therapy. In recent years, progress in understanding the genetic and molecular alterations underlying sarcomas has led to improved diagnostic techniques and therapeutic strategies.1,2
Around 80 per cent of sarcomas are classed as soft tissue sarcomas (STSs), 15 per cent as bone sarcomas, and 5 per cent as gastro-intestinal stromal tumours (GISTs).3, 4 Bone sarcomas are more common among children, while STSs are more common in adults.9 Approximately 220 people are diagnosed annually in Ireland with some type of sarcoma.8
STSs are a group of more than 60 different neoplasms that can originate from any location throughout the human body. They can span a range of clinical presentations, from aggressive metastatic angiosarcomas to benign lipomas.7 More than half of STSs are found in the leg. Globally, the incidence of STS is around three-to-four per 100,000 annually, which accounts for 1 per cent of all adult solid malignant tumours and more than 20 per cent of paediatric cancers.5
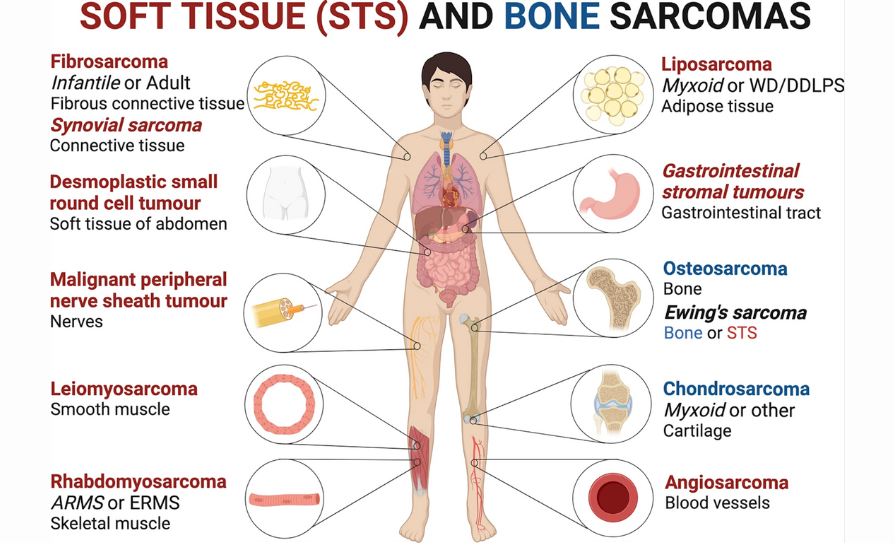
FIGURE 1: Schematic representation of the most frequently occurring soft tissue (STS) (red) and bone (blue) sarcomas and affected tissues. Sarcomas with a simple karyotype are referred to in italics: ARMS alveolar rhabdomyosarcoma; ERMS
embryonal rhabdomyosarcoma; WD/DDLPS well-differentiated/dedifferentiated liposarcoma.
Source: www.nature.com/articles/s41392-021-00647-8
Types of STS
Liposarcoma is the most common STS. They are most common in adults between 30 and 60 years of age and are slightly more common in men. These tumours usually develop in the deep fatty tissue and are most often found in the upper leg, behind the knee, the groin, the buttock area, or behind the organs in the abdominal cavity.
There are different types of liposarcoma, which include well differentiated liposarcoma, usually a large tumour in the deep tissues of the abdominal cavity, mediastinum, and groin region; dedifferentiated liposarcoma, found in similar areas to differentiated liposarcoma but the tumour is of higher grade; pleomorphic liposarcoma (high grade), most commonly found on the lower limbs and feet; and myxoid liposarcoma, most commonly seen in the deep soft tissue of limbs, especially behind the knee.
Well differentiated liposarcoma and dedifferentiated liposarcoma are associated with a particular gene alteration involving chromosome 12. Myxoid liposarcoma is associated with an alteration of the DDIT3 gene. There is currently no known recurrent gene alteration associated with pleomorphic liposarcoma.8
Myxofibrosarcomas are sarcomas in fibrous tissue such as muscles, nerves, and blood vessels. They are usually found in the arms or legs, or on the trunk of the body, but can occur in any fibrous tissue, including scars, muscles, nerves, tendons, and around the lining of the bone. There is currently no known recurrent specific gene alteration associated with myxofibrosarcoma.8
Dermatofibrosarcoma protuberans (DFSP) is a sarcoma of superficial soft tissues, usually involving the skin. It is most common in people aged between 20 and 50, although a small percentage can occur in children. Most commonly it affects the head and neck area, trunk, upper arms, and legs. DFSP is characteristically associated with a particular gene alteration involving the COL1A1 gene.8
Synovial sarcoma generally occurs in young adults and is mainly found in the arms or legs next to a joint. These sarcomas are usually found around the joint capsule, but rarely spread into the joint itself. The most common site is next to the knee, as well as the foot, ankle, and hand. Unlike other STSs, synovial sarcomas are frequently painful. Nearly all synovial sarcomas are associated with an alteration in the SS18 gene. This can help to confirm a diagnosis.8
Epithelioid sarcoma is divided into classical and proximal types. Classical epithelioid sarcomasare usually found in the hand or foot of young adults. Proximal type epithelioid sarcoma is usually found on the trunk and upper arm and leg areas. They appear like small lumps, which sometimes co-join. The cancer spreads to the lymph nodes in about one-in-five cases.8
Perivascular epithelioid cell tumours (PEcoma) are a rare type of soft tissue tumour which may be found in the lung, intra-abdominal cavity, or female genital tract. Most of these tumours are benign, but some show malignant pathological features and behave more aggressively.8
Rhabdomyosarcoma refers to a striated muscle tumour. About half of all STSs found in children are rhabdomyosarcoma. There are many different types, including embryonal, alveolar, botryoid, and pleomorphic.8
Leiomyosarcomas are another type of muscle tumour which usually occur in the leg, bowel, or uterus. Symptoms of bowel or uterine leiomyosarcomas are bleeding and pain.8
Gastro-intestinal stromal tumours (GISTs) develop in tissue around the stomach and intestines, and are relatively common. They are usually treated with surgery alone, but some people require drug treatment before or after removal.8
Mixoid STSs are generally subclassified as intramuscular myxomas, juxta-articular myxomas, and cutaneous myxomas. They occur in men and women around the age of 50, and are most often found in the arms and legs. Tumours can be small lumps or very large tumours. They are benign and do not spread to other parts of the body; however, some subtypes can spread locally to tissue close to the tumour.8
Solitary fibrous tumour (SFT) is a soft tissue tumour that can affect any site in the body including the pleura, soft tissues of the limbs, trunk, and within the abdominal cavity. Most SFTs are benign, but some display more aggressive pathological features and have the potential to spread to other parts of the body. SFTs are associated with a particular gene alteration termed a ‘NAB2-STAT6 gene fusion’, which is detected in the vast majority of SFT cases.8
Mesenchymomas can be distributed throughout the body and about three-out-of-four are malignant. The tumours are invasive, especially when they are in the skeletal muscle. Treatment is wide excision surgery, sometimes combined with radiation and or chemotherapy.8
The vascular sarcomas epithelioid haemangioendotheliomas are very rare tumours of the blood vessels, which can be cancerous. They are found in men and women, but rarely in children. They may manifest in the skin, liver, or bone. About a third of cases occur on the skin and a quarter are found in the soft tissue of organs such as breast, liver, heart, and lungs.8
Kaposi sarcoma is a vascular tumour that has different clinical subtypes. The classical type arises within the skin, especially over the lower legs in adults. A second type can affect children or adults and may involve lymph nodes. A third type affect patients with retroviral infection and may involve skin, lungs, the digestive tract, and the mouth. All variants of Kaposi sarcoma are associated with human herpes virus-8 (HHV8).8
Malignant peripheral nerve sheath tumours are usually found in young-to-middle-aged adults and are more common in males. They occur in nerves outside the brain or spinal cord. These tumours spread to the surrounding soft tissue forming a lumpy tumour. This type of cancer can spread through the bloodstream. They can be painful and tender, and may occur in patients who have neurofibromatosis.8
Alveolar soft part sarcoma is a very rare tumour that occurs in adolescents and young adults. It is usually a slow-growing tumour found in the arms and legs. It is associated with an alteration in the TFE3 gene.8
Desmoid tumours are slow-growing soft tissue sarcomas that tend to spread to nearby tissues, but not to other parts of the body. Desmoid tumours may occur sporadically (sporadic desmoid) or may occur in association with an underlying genetic condition known as familial adenomatous polyposis (FAP) syndrome (hereditary desmoid). Patients with this syndrome may also be at risk for tumours in the lower digestive tract.8
Types of bone sarcoma
There are several different types of bone sarcoma, and like STSs, most are named after the cells in which they grow.
Definitions of TNM
Primary Tumour (T)
| TX | Primary tumour cannot be assessed |
| T0 | No evidence of primary tumour |
| T1 | Tumour 5cm or less in greatest dimension |
| T1a | Superficial tumour |
| T1b | Deep tumour |
| T2 | Tumour more than 5cm in greatest dimension |
| T2a | Superficial tumour |
| T2b | Deep tumour |
Note: Superficial tumour is located exclusively above the superficial fascia without invasion of the fascia; deep tumour is located either exclusively beneath the superficial fascia, superficial to the fascia with invasion of or through the fascia, or both superficial yet beneath the fascia.
Regional Lymph Nodes (N)
| _NX | Regional lymph nodes cannot be assessed |
| N0 | No regional lymph node metastasis |
| N1* | Regional lymph node metastasis |
*Presence of positive nodes (N1) in M0 tumours is considered Stage III
Distant Metastasis (M)
| M0 | No distant metastasis |
| M1 | Distant metastasis |
Anatomic Stage / Prognostic Groups
| GROUP | T | N | M | Grade |
|---|---|---|---|---|
| IA | T1a T1b | N0 N0 | M0 M0 | G1, GX G1, GX |
| IB | T2a T2b T1a T1b | N0 N0 N0 N0 | M0 M0 M0 M0 | G1, GX G1, GX G1, GX G1, GX |
| IIA | T1a T1b | N0 N0 | M0 M0 | G2, G3 G2, G3 |
| IIB | T2a T2b | N0 N0 | M0 M0 | G2 G2 |
| III | T2a, T2b Any T | N0, N1 | M0 | G3 Any G |
| IV | Any T | Any N | M1 | Any G |
FIGURE 2: TNIM staging of soft tissue sarcoma. Source: www.cancertherapyadvisor.com/home/decision-support-in-medicine/oncology/bone-andsoft-tissue-sarcomas/ www.openaccessjournals.com/articles/fdg-petct-as-a-marker-for-gradingsarcomas-and-for-the-individualization-of-disease-management-9426.html
Ewing’s sarcoma encompasses several types of sarcomas known as the Ewing family of tumours. In Ireland, there are about 20 cases diagnosed each year, usually in children and young adults under the age of 30. It can be found in any bone, but is most common in the bones of the lower body such as the pelvis, tibia, fibula, and femur. The vast majority of Ewing’s sarcoma cases are associated with an alteration in the EWSR1 gene, which is helpful in confirming the diagnosis on core biopsy.8 BCOR associated sarcoma is like Ewing’s sarcoma but the gene alteration with this type of tumour affects the BCOR gene.8
Chondrosarcoma develops from cartilage-producing cells. Less than one-third of bone sarcomas are chondrosarcomas, and it is usually found in older people. Unlike other bone cancers, chondrosarcoma is more often found in the spine and pelvis than in the arms or legs.8
Osteosarcoma mainly occurs in adolescents and young adults. It usually affects the humerus of the upper arm and the femur and tibia of the leg. In children and adolescents, 80 per cent of these tumours arise from the bones around the knee. It is slightly more common in males than females. Although osteosarcoma is a common malignant bone tumour, it is still rare with fewer than 30 new cases annually in Ireland.8
Parosteal osteosarcoma is a slow growing low-grade tumour that grows on the surface of the bone. It typically occurs in adults between the ages of 20 and 40 and it is mainly found on the upper leg, behind the knee.8 Periosteal osteosarcoma is an uncommon tumour that grows on the surface of the bone — usually the lower leg. It is a fast-growing high-grade tumour and occurs in younger people.8
Multifocal sclerosing osteosarcoma is an extremely rare form of osteosarcoma that tends to occur in children under 10.8 Osteosarcoma of the jaw and skull is also rare, and typically occurs in people between the ages of 20 and 40. The bones most often affected are the mandible and the maxilla.8
Osteosarcoma in Paget’s disease usually occurs in people over the age of 60. About one-in-100 patients with Paget’s disease develop primary bone sarcoma. The tumours usually start in the pelvis, femur, or humerus.8
Radiation-induced osteosarcoma is a rare form of osteosarcoma which occurs in people who have undergone radiation treatment for other cancers. These tumours appear about 10 years after radiation therapy. Post-irradiation osteosarcomas are mainly found in the spine, pelvis, hips, and shoulders. 8 Extra-osseous sarcomas develop away from the bone and are usually found in the muscle or skin.
There are three types — giant cell tumours, osteosarcomas, and Ewing’s sarcoma. 8
Risk factors
Most sarcomas do not have a known cause, although there are several factors that could increase the risk of development. Patients who have received radiation therapy for previous cancers may have a higher risk.
People with a family history of inherited disorders, such as Von Recklinghausen’s disease (neurofibromatosis), Gardner syndrome, Werner syndrome, tuberous sclerosis, naevoid basal cell carcinoma syndrome, Li-Fraumeni syndrome, or retinoblastoma, have a higher risk of developing a sarcoma.
Exposure to vinyl chloride monomer — a chemical substance used to make some types of plastics — dioxin, or arsenic may also increase the risk. However, most sarcomas are not known to be associated with specific environmental hazards. Having lymphoedema in the arms or legs for a long time may also increase the risk of developing a sarcoma.9
Symptoms
Neoplasms that are small, superficial, and mobile are highly suggestive of STS. STSs present as a lump, usually more than 4-to-5cms in diameter, deep in the body tissues underneath the skin. They are not painful to touch, but pain may occur as the lump gets bigger.
Several conditions can mimic STS, including hypertrophic scars, haematoma, benign lipoma, cysts, abscesses, and melanoma. Symptoms of bone sarcomas include a hard growing lump and pain around a bone, a limited range of motion in a joint, or a bone that breaks without an apparent cause.8, 9
Diagnosis and evaluation
Diagnosis is based on symptoms, medical history, clinical examination, and tests including x-ray, blood tests, MRI, CT scan, ultrasound, PET scan, biopsy, core needle biopsy, and surgical biopsy. Indications for preoperative imaging and biopsy consider the extent of the mass on physical examination, and the anticipated neurovascular involvement.
The likelihood of nodal involvement or distant metastases must be considered as well as the relative resectability and potential postoperative functional deficits. MRI is generally considered the most informative for trunk and extremity STS. Chest CT with contrast is considered in cases with high metastatic potential, as the lungs are often involved.
The use of PET/CT in the workup of sarcomas has not yet become the standard of care. If a biopsy is recommended, then the choice is a core-needle biopsy, and if this is nondiagnostic, then an incisional biopsy may need to take place.7
Staging
Staging of STSs utilises the TNM [tumour, node, metastasis] system as well as tumour grading, as outlined in Figure 2.
Treatment
Sarcoma is treated with a combination of chemotherapy, radiation, and surgery. Treatment plans and recovery depend on a variety of factors, including the type of sarcoma, the tumour location, grade and size, patient’s age, and whether the cancer is new or recurrent.8, 9 Surgical resection is a mainstay of sarcoma treatment. Patients that are operative candidates are encouraged to undergo resection.7
Treatment for liposarcoma is surgical resection with wide margins. Local recurrence is common. For liposarcomas of the extremities, the goal of care is limb-sparing resection with a gross negative margin. For retroperitoneal liposarcomas, the goal is complete resection. Typically, well-differentiated liposarcomas have a low risk of distant metastases, whereas de-differentiated extremity liposarcoma generally benefits from neoadjuvant radiation therapy.7
The modern surgical approach to sarcoma resection consists of a wide en-bloc resection with the goal of at least a 1cm margin of uninvolved tissue in all directions. A 2cm margin may be considered for tumours with infiltrative borders. However, this may not be possible due to the extent of the disease or proximity to neurovascular structures.
However, if the tumour is close to or displacing neurovascular structures, they do not need to be resected if the adventitia and/or perineurium are removed. While bone is rarely involved, it may need to be resected to obtain adequate margins. A planned positive margin followed by adjuvant therapy may be required.7
For locally recurrent tumours, surgery may still be an option with either a wide excision or amputation. The decision to operate, and type of operation, is based on location, size, proximity to vital structures, and ability to preserve limb function. Tumours that are large and unresectable, or present in the patient not considered a surgical candidate, may be observed if there are no symptoms. In symptomatic individuals, palliative surgery may be offered.7
Surgery alone in the case of high-grade sarcomas has a recurrence rate of 33 per cent at five years, and adjuvant radiotherapy is typically recommended. Radiotherapy can be incorporated into any phase of treatment for STS of the trunk and extremities, ie, pre-operatively, post-operatively, intraoperatively, and concurrently with chemotherapy.
Indications for radiotherapy include high-grade stage II+ inoperable disease, recurrent disease, or positive margins. The two most used techniques are external beam radiotherapy and brachytherapy. External beam radiotherapy is the most common means of treating sarcomas, and requires the patient to undergo a CT simulation.
Pre-operative radiotherapy is the preferred method because of several advantages. It allows for lower doses of radiation, easier target delineation, ease of set-up, smaller radiation field size, and better functional outcomes with similar rates of local control. However, adjuvant therapy may be needed in the case of positive margins or a high-grade tumour where pre-operative treatment was not planned.
Postoperative radiotherapy is more challenging from a planning perspective due to the surgical manipulation of the tissue. Pre and postoperative imaging are required and should be incorporated into treatment planning.7
Brachytherapy delivers radiation precisely to the target tissue with high conformity, sparing the nearby normal tissues and eliminating the possibility of normal tissue toxicities. Brachytherapy is typically utilised post-operatively as either monotherapy combined with external beam radiotherapy as a boost, as well as for recurrent disease. It may be delivered at a high dose rate (HDR), low dose rate (LDR), or pulsed dose rate (PDR). 7, 10
Chemotherapy uses anti-cancer drugs such as doxorubicin and ifosfamide to destroy cancer cells. It is most frequently used if the sarcoma has spread to other parts of the body and is also used for sarcomas such as osteosarcoma, Ewing’s sarcoma, and rhabdomyosarcoma.11 Chemotherapy can also be given before or after radiotherapy and surgery, or in adjunct with radiotherapy, to improve radiotherapy outcomes.8
Prognosis
Despite the heterogeneity of sarcomas, the most important prognostic factors are histologic grade and tumour size. Histologic grade predicts distant metastasis and survival, while primary tumour size predicts local recurrence and distant metastasis. In general, for localised and early-stage lesions, curative resection can be done with good long-term survival, but recurrences are common.
For those with advanced disease, a cure is not possible, and the median survival is 12-to-18 months, depending on the subtype. The risk of recurrence persists even after 10-to-15 years, and patients need indefinite follow-up. The majority of recurrences occur within the first five years. Most sarcomas show a poor response (10-to-50 per cent) to chemotherapy. Again, the response also depends on histological subtype, grade, and patient.7
Future directions and challenges
Novel therapeutic modalities, such as antibody-drug conjugates, bispecific T-cell engagers (BiTEs), and chimeric antigen receptor (CAR) T-cell therapy, hold potential in the treatment of sarcomas. These innovative approaches are being investigated in preclinical and early-phase clinical trials.
Despite much progress, several challenges remain in the field of sarcoma research and management, including the need for larger clinical trials, identification of biomarkers for treatment response, and optimisation of multimodal treatment strategies. Collaboration among researchers, clinicians, and patient advocacy groups is paramount in addressing these challenges and improving outcomes for sarcoma patients.1,6
References
- Damerell V, Pepper M, Prince S. Molecular mechanisms underpinning sarcomas and implications for current and future therapy. Sig Transduct Target Ther 2021;6:246.
- Fletcher C, Bridge J, Hogendoorn P, Mertens F. WHO classification of tumours of soft tissue and bone. 4th Edition, Volume 5. Lyon: International Agency for Research on Cancer Press; 2013.
- Anttila S, Boffetta P. Occupational cancers. USA: Springer; 2020.
- Zhao X, Yue C. Gastrointestinal stromal tumor. J Gastrointest Oncol. 2012;3(3):189-208.
- Bray F, Colombet M, Mery L, et al. Cancer incidence in five continents. Lyon: International Agency for Research on Cancer; 2017.
- Dickson M, et al. New strategies in soft tissue sarcoma: The role of immunotherapy. Clin Cancer Res. 2022;28(7):1583-1591.
- Popovich J, Kashyap S, Gasalberti D, et al. Sarcoma. StatPearls Publishing. 2023. Available from www.ncbi.nlm.nih.gov/books/NBK519533/.
- Irish Cancer Society. Sarcoma. 2023. Available from www.cancer.ie/cancer-information-and-support/cancer-types/sarcoma.
- John Hopkins Medicine. Sarcoma. 2023. Available from www.hopkinsmedicine.org/health/conditions-and-diseases/sarcoma.
- Manir KS, Basu A, Choudhury KB, et al. Interstitial brachytherapy in soft tissue sarcoma: a 5 years institutional experience with Cobalt 60-based high-dose-rate brachytherapy system. J Contemp Brachytherapy. 2018;10(5):431-438.
- Lee, B. Bone and soft tissue sarcoma. Cancer Therapy Advisor. 2023. Available from www.cancertherapyadvisor.com/ddi/bone-cancer-pharmacologic-treatment/.