







Immunoglobulin A nephropathy (IgAN), also known as Berger’s disease, is the commonest form of primary glomerulonephritis (GN) and is a major global cause of chronic kidney disease (CKD).1,2 Subsequent renal failure occurs in 20-to-40 per cent of patients with IgAN within 10-to-20 years from diagnosis.3
The condition may affect people of almost all ages; however, most patients are diagnosed before the age of 40,4 with peak incidence in the second and third decades of life.5 Diagnosis is rare in children under five years of age.6 In countries in which IgAN is commonly found, life expectancy varies between 70-to-85 years.7
There is a substantial variation in incidence of IgAN between ethnic/racial groups, with the highest incidence being in the East Asian population, followed by Europeans.8 The male-to-female ratio has been stated to be 2-3:1 in Europeans9 and North Americans10 and 1:1 in East Asians.11
Risk factors and associated conditions
Apart from being in the 20-to-30 age group, having an East Asian ethnicity, and a male gender, other risk factors which have been implicated in the development of IgAN include susceptibility to the common cold, a familial history of chronic nephritis, frequent consumption of raw eggs, carbohydrates, and salty foods.12 High levels of triglycerides and uric acid have also been mentioned as risk factors for progression of IgAN.13
Various systemic conditions are associated with the development of histological
and clinical manifestations of IgAN. These include:
- Gastrointestinal and liver diseases (eg, coeliac disease, inflammatory bowel disease);
- Autoimmune diseases (eg, rheumatoid arthritis, ankylosing spondylitis);
- Infection (eg, hepatitis B and C, human immunodeficiency virus);
- Skin conditions (eg, psoriasis, dermatitis herpetiformis);
- Respiratory tract conditions (eg, cystic fibrosis, sarcoidosis);
- Malignancy (eg, renal cell wcarcinoma, lung cancer).3
Clinical presentation
IgAN can follow various courses, ranging from asymptomatic abnormalities of the urinary system with potential to resolve spontaneously, to rapidly progressive GN (RPGN) with renal failure.14 In the paediatric and adolescent populations, painless macroscopic haematuria, often in conjunction with an upper respiratory or gastrointestinal tract infection, is the typical initial clinical picture.6 Macroscopic haematuria across all ages may also be provoked by extreme physical activity.6 Most individuals with macroscopic haematuria, at any age, have repetitive episodes over a number of years.15
In the adult population, asymptomatic microscopic haematuria, with or without proteinuria and/or progressive kidney disease, is the most common presentation, and may be detected during routine health screenings.3
The urine is often brown rather than red, commonly described as ‘cola-coloured’, and the passage of blood clots is rare. Bilateral loin pain is occasionally present during these episodes, likely due to oedema of the renal capsule.16 The degree of proteinuria in such patients varies widely amongst individuals, although proteinuria in the absence of microscopic haematuria is rare.38
Another presentation of adult IgAN is with synpharyngitic macroscopic haematuria, where the patient develops gross haematuria along with an upper respiratory tract infection, or less likely, gastrointestinal tract infections. In such cases, haematuria is usually visible at the same time as infective symptoms, hence the term ‘synpharyngitic’ in comparison with the two-to-three-week gap seen between the onset of infection and gross haematuria in patients with post-infectious GN (post-pharyngitic).
This presentation occurs in only 10-to-15 per cent of adult patients with IgAN, and these are typically individuals aged less than 40. This phenotype is commonly associated with a favourable prognosis.3
RPGN is a clinical syndrome characterised by a decline in estimated glomerular filtration rate (eGFR) by ≥50 per cent over three months or less in cases where reversible causes have been excluded. This can occur either at presentation of the disease or during the course of IgAN, and may present without any clinical features of IgA vasculitis. RPGN also displays poor kidney prognosis.3
Nephrotic syndrome is a rare presentation of IgAN seen only in ≤5 per cent of cases40 and it develops more often in children than in adults.16 Patients presenting with the clinical features of nephrotic syndrome (including hypoalbuminaemia) cannot be distinguished from those presenting with minimal change disease.
Kidney biopsy shows mesangial IgA deposition along with extensive foot process effacement with or without mesangial cell proliferation. Given that this clinical entity is rare, it should be distinguished from the more common presentation of ‘nephrotic-range’ proteinuria, which is seen in some patients with IgAN who do not typically have hypoalbuminaemia.3
Acute kidney injury (AKI) is uncommon in IgAN, occurring in ≤5 per cent of cases. When present, its development is described by two distinct mechanisms. The first is an acute, severe, inflammatory damage of capillaries located in the glomerular tuft which leads to leakage of circulatory proteins in the urinary space, subsequently inducing proliferation of epithelial cells in the Bowman’s capsule forming crescents.16 Alternatively, AKI may present with minimal glomerular injury when substantial haematuria causes damage to tubular epithelial cells or tubular obstruction by red cell casts.17
Decline in kidney function in IgAN is usually chronically progressive.3 IgAN is a common cause of CKD, especially in patients with persistent proteinuria more than 1g/day.18
Classification
A commonly used classification system is the Oxford Classification of IgAN.19 This system defines five prognostic features which are scored subjectively in renal biopsies and include:
- Mesangial cellularity (M): Defined as ≥4 mesangial cells in any mesangial area of a glomerulus.
- Endocapillary hypercellularity (E): Defined as an increased number of cells in the glomerular capillary lumen.
- Segmental sclerosis (S): Defined as adhesion or sclerosis which is not involving the entire glomerulus.
- Interstitial fibrosis/tubular atrophy (T): Defined as the percentage of tubular atrophy and/or interstitial fibrosis of cortical area.
- (fibro) Cellular crescents (C): Defined as extracapillary cell proliferation more than two cell layers in thickness and <50 per cent matrix.20,21
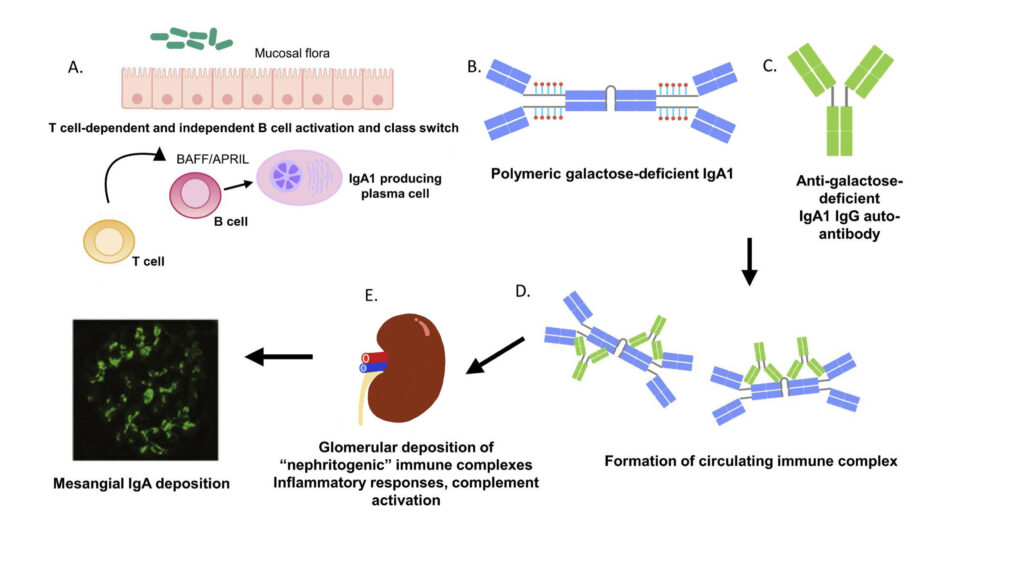
FIGURE 1: The pathogenesis of IgAN as described using the multi-hit hypothesis31
Diagnosis
There is no solitary or combined laboratory test that can diagnose IgAN; the final diagnosis requires histological examination of kidney tissue. Preliminary tests are carried out in order to determine the need for kidney biopsy.22
Urinalysis and quantification of proteinuria: Careful examination of a freshly voided first morning urine sample using unstained bright field microscopy or phase contrast microscopy is useful as an initial tool to diagnose IgAN. Glomerular bleeding is indicated by the presence of red blood cell casts and dysmorphic red blood cells.
However, this finding also occurs in thin basement membrane lesions (benign familial haematuria) and hereditary nephritis (X-linked Alport syndrome). A timed urine collection or a spot urine protein-to-creatinine ratio measurement enables quantification of proteinuria. Serum complement values are usually normal but may sometimes be decreased.22
Other urine biomarkers: A fraction of GdIgA1 (alactose deficient IgA1) from glomerular deposits is excreted in urine and can therefore potentially be used as a disease-specific biomarker of IgAN.23 Increased urinary levels of cytokines and/or growth factors are also observed in patients with IgAN.24
Renal function: The assessment of renal function by endogenous creatinine clearance and eGFR at baseline, and serially six monthly after that, for eGFR is of utmost importance. This is because CKD at baseline has valuable prognostic implications.22
Serum biomrkers: Serum total IgA levels are increased in approximately 30-to-50 per cent of patients with IgAN. However, this is neither a specific nor a sensitive biomarker for the condition. These levels bear no prognostic value when it comes to IgAN.22 GdIgA1 and the corresponding auto-antibodies that detect it are the most widely investigated serum biomarkers.25
Various studies have reported increased levels of GdIgA1 in the circulation of IgAN patients compared to other renal diseases and healthy individuals in multiple populations.26 Another potential serum biomarker could be miRNAs which regulate gene expression. The presence of these molecules might be able to differentiate patients with primary IgAN from patients suffering from other forms of primary GN.27
Histological examination: Routine immunofluorescence microscopy shows IgA as the predominant or codominant immunoglobulin in the glomerular immune deposits.6 Distinctive characteristics attributed to this IgA include restriction to the IgA1 subclass,28 and it contains less galactose in its O-glycans than does circulating IgA1 in healthy individuals.29
The immune proteins in the glomeruli of IgAN patients usually include complement C3; and IgG, IgM, or both are often also present.5 The typical findings with light microscopy would be glomerular injury appearing as increased mesangial matrix and mesangial hypercellularity.30
Pathogenesis
The pathogenesis of IgAN is commonly described using the multi-hit hypothesis (Figure 1). IgA-producing plasma cells located in the mucosa-associated lymphoid tissue (MALT) produce pathogenic IgA. There is activation of B-cells which, in turn, undergo class switching via both T-cell dependent and independent mechanisms.
BAFF (B-cell activating factor) and APRIL (a proliferation-inducing ligand), both tumour necrosis factor-related cytokines, play a central role in T-cell-independent induction of B-cells and class switching to Ig-A-producing plasma cells. Gd-IgA1 are found at increased levels in patients with IgAN and these aggregate to form immune complexes which are detected in the kidneys and circulation. These immune complexes may subsequently activate complement and trigger inflammatory responses inside the glomerulus.31
Treatment
Supportive therapy should prioritise blood pressure management, and target sitting systolic blood pressure should be <120mmHg. Preferred antihypertensives include angiotensin converting enzyme inhibitors (ACEi) and angiotensin receptor blockers (ARBs) as first choice, with dosage up-titration as tolerated.
These should be given to all patients with proteinuria of more than 5g/d and there should be no combination therapy. Other antihypertensives which may be used include non-dihydropyridine calcium channel blockers (eg, verapamil), aldosterone antagonists, and beta-blockers. Dihydropyridine calcium channel blockers, such as amlodipine, should be avoided.14
Statin therapy should also be initiated if persistent hyperlipidemia is present.3 3-Hydroxy-3-methylglutaryl–coenzyme A (HMG-CoA) reductase inhibitors may be used in such cases as these have a reno-protective effect.32
Dietary advice and fluid balance are also important. Sodium intake should be restricted to less than 2g per day, while protein intake should be controlled, as should fluid intake to less than 1.5-to-2 litres per day.14 Lifestyle modifications include smoking cessation, normalisation of body weight, and regular endurance sports with avoidance of vigorous exercise.14
Additional measures include avoidance of non-steroidal anti-inflammatory drugs and prolonged severe hyperkalaemia.
The use of hydroxychloroquine in patients with proteinuria despite maximum dosage of renin-angiotensin-system (RAS) blockers, and the use of sodium-glucose cotransporter 2 inhibitors, may be considered.
Immunosuppressive therapy: Patients who remain at high risk of progression to kidney failure, that is, patients with proteinuria of ≥0.75-1g/d despite three months of optimal, supportive care should ideally be enrolled in an IgAN-focused clinical trial. If this is unavailable or not feasible, then immunosuppressive should be considered.33 However, this should involve careful consideration of the patient characteristics and preferences along with the expected risks and benefits.34
Corticosteroids are currently the most commonly used immunosuppressive therapy for IgAN.34 Apart from immunosuppressive properties, these drugs also have anti-inflammatory properties.35
According to the 2021 KDIGO (Kidney Disease: Improving Global Outcomes) Glomerular Guidelines,35 the clinical benefit of glucocorticoids has not been established, hence these drugs should be given with extreme caution and avoided entirely in patients with obesity, diabetes, an eGFR <30ml/min/1.73m2, latent infections (eg, tuberculosis), secondary disease (eg, cirrhosis), severe osteoporosis, uncontrolled psychiatric illnesses, and active peptic ulceration.
These guidelines also suggest that, when appropriate, treatment with glucocorticoid (prednisolone equivalent ≥0.5mg/kg/d) should include prophylaxis against Pneumocystis pneumonia along with gastro and bone protection as per local guidelines.
Alternative immunosuppressive methods: In Chinese patients, mycophenolate mofetil (MMF) at 1.5g/d along with low-dose prednisolone for six months offers an effective and potentially safer alternative to systemic corticosteroids. However, this still carries an increased risk of complications.
The use of hydroxychloroquine, which is thought to have pleiotropic immunomodulatory effects, can also be considered as an alternative option for Chinese patients.34 Tonsillectomy with concurrent corticosteroid therapy may be considered in Japanese patients as this has been associated with a reduction in proteinuria and improved kidney survival rates in these populations.3,34
Experimental therapies: Targeted corticosteroid delivery to the ileum is a novel approach which reduces systemic toxicity whilst achieving the required efficacy.34 The rationale behind this treatment is to release targeted-release formula of budesonide at the distal ileum, which is the location of the largest area of Gd-IgA1, specifically MALT. Several complement inhibitors are being studied for the management of IgAN including narsoplimab, LNP023, iptacopan, pegcetacoplan, cemdisiran, and avacopan amongst others.37
Renal replacement therapy (RRT): In IgAN patients who require RRT, transplantation is the method of choice.16 However, IgAN does unfortunately frequently recur in the allograft, with a recurrence rate of about 50 per cent at 10 years.38 Research by Uffing et al39 proposed a 3.7-fold increased risk of post-transplant graft loss in patients with IgA nephropathy.
Conclusion
IgAN is one of the most common GNs and its clinical presentation varies drastically among patients. The early diagnosis of IgAN is of utmost importance as delaying diagnosis and appropriate management can have an impact on the preservation of the patient’s renal function and overall outcome. Favourable outcomes in such patients depend on early detection and timely treatment.
References
- Feltis JT, Churg J, Holley KM, et al. Active and chronic phases of Berger’s disease (IgA nephropathy). Am J Kidney Dis. 1984;3(5):349-356.
- Canney M, Barbour SJ, Zheng Y, et al. Quantifying duration of proteinuria remission and association with clinical outcome in IgA nephropathy. J Am Soc Nephrol. 2021;32(2):436-447.
- Pattrapornpisut P, Avila-Casado C, Reich HN. IgA nephropathy: Core curriculum 2021. Am J Kidney Dis. 2021;78(3):429-441.
- Jarrick S, Lundberg S, Welander A, et al. Mortality in IgA nephropathy: A nationwide population-based cohort study. J Am Soc Nephrol. 2019;30(5):866-876.
- Wyatt RJ, Julian BA. IGA nephropathy. New Eng J Med. 2013;368(25):2402-14.
- Knoppova B, Reily C, King RG, et al. Pathogenesis of IgA nephropathy: Current understanding and implications for development of disease-specific treatment. J Clin Med. 2021;10(19):4501.
- World Health Organisation. Life expectancy and healthy life expectancy by country. Global Health Observatory Data Repository. Geneva: WHO; 2020. Available at: www.who.int/data/gho/data/themes/mortality-and-global-health-estimates/ghe-life-expectancy-and-healthy-life-expectancy.
- Woo KT, Lau YK, Chan CM, et al. Angiotensin-converting enzyme inhibitor versus angiotensin 2 receptor antagonist therapy and the influence of angiotensin-converting enzyme gene polymorphism in IgA nephritis. Ann Acad Med Singap. 2008;37(5):372-376.
- Geddes CC, Rauta V, Gronhagen-Riska C, et al. A tricontinental view of IgA nephropathy. Nephrol Dial Transplant. 2003;18(8):1541-1548.
- Wyatt RJ, Kritchevsky SB, Woodford SY, et al. IgA nephropathy: Long-term prognosis for paediatric patients. J Pediatr. 1995;127(6):913-919.
- Shen AYJ, Brar SS, Khan SS, et al. Association of race, heart failure, and chronic kidney disease. Future Cardiology. 2006;2(4):441-54.
- Wakai K, Kawamura T, Matsuo S, et al. Risk factors for IgA nephropathy: A case-control study in Japan. Am J Kidney Dis. 1999;33(4):738-745.
- Syrjänen J, Mustonen J, Pasternack A. Hypertriglyceridaemia and hyperuricaemia are risk factors for progression of IgA nephropathy. Nephrol Dial Transplant. 2000;15(1):34-42.
- Floege J, Rauen T, Tang SCW. Current treatment of IgA nephropathy. Semin Immunopathol. 2021;43(5):717-728.
- Radford MG Jr, Donadio JV Jr, Bergstralh EJ, et al. Predicting renal outcome in IgA nephropathy. J Am Soc Nephrol. 1997;8(2):199-207.
- Rajasekaran A, Julian BA, Rizk DV. IgA nephropathy: An interesting autoimmune kidney disease. Am J Med Sci. 2021;361(2):176-194.
- Gutiérrez E, González E, Hernández E, et al. Factors that determine an incomplete recovery of renal function in macrohematuria-induced acute renal failure of IgA nephropathy. Clin J Am Soc Nephrol. 2007;2(1):51-57.
- Reich HN, Troyanov S, Scholey JW, et al. Remission of proteinuria improves prognosis in IgA nephropathy. J Am Soc Nephrol. 2007;18(12):3177-3183.
- Trimarchi H, Barratt J, Cattran DC, et al. Oxford Classification of IgA nephropathy 2016: An update from the IgA Nephropathy Classification Working Group. Kidney Int. 2017;91(5):1014-1021.
- Markowitz G. Glomerular disease: Updated Oxford classification of IgA nephropathy: A new MEST-C score. Nat Rev Nephrol. 2017;13(7):385-386.
- Howie AJ, Lalayiannis AD. Systematic review of the Oxford classification of IgA nephropathy: Reproducibility and prognostic value. Kidney360. 2023;4(8):1103-1111.
- Lai KN, Tang SC, Schena FP, et al. IgA nephropathy. Nat Rev Dis Primers. 2016;2:16001.
- Serino G, Pesce F, Sallustio F, et al. In a retrospective international study, circulating miR-148b and let-7b were found to be serum markers for detecting primary IgA nephropathy. Kidney Int. 2016;89(3):683-692.
- Suzuki H, Allegri L, Suzuki Y, et al. Galactose-deficient IgA1 as a candidate urinary polypeptide marker of IgA nephropathy? Dis Markers. 2016;2016:7806438.
- Yang YZ, Liu LJ, Shi SF, et al. Effects of hydroxychloroquine on proteinuria in immunoglobulin A nephropathy. Am J Nephrol. 2018;47(3):145-152.
- Vaz de Castro PAS, Amaral AA, Almeida MG, et al. Examining the association between serum galactose-deficient IgA1 and primary IgA nephropathy: A systematic review and meta-analysis. J Nephrol. Published online March 1, 2024.
- Yanagawa H, Suzuki H, Suzuki Y, et al. A panel of serum biomarkers differentiates IgA nephropathy from other renal diseases. PLoS One. 2014;9(5):e98081.
- Conley ME, Cooper MD, Michael AF. Selective deposition of immunoglobulin A1 in immunoglobulin A nephropathy, anaphylactoid purpura nephritis, and systemic lupus erythematosus. J Clin Invest. 1980;66(6):1432-1436.
- Allen AC, Bailey EM, Brenchley PE, et al. Mesangial IgA1 in IgA nephropathy exhibits aberrant O-glycosylation: Observations in three patients. Kidney Int. 2001;60(3):969-973.
- Roberts IS. Pathology of IgA nephropathy. Nat Rev Nephrol. 2014;10(8):445-454.
- Suzuki H, Kiryluk K, Novak J, et al. The pathophysiology of IgA nephropathy. J Am Soc Nephrol. 2011;22(10):1795-1803.
- Moriyama T, Oshima Y, Tanaka K, et al. Statins stabilise the renal function of IgA nephropathy. Ren Fail. 2014;36(3):356-360.
- Rovin BH, Adler SG, Barratt J, et al. Executive summary of the KDIGO 2021 guideline for the management of glomerular diseases. Kidney Int. 2021;100(4):753-779.
- Gleeson PJ, O’Shaughnessy MM, Barratt J. IgA nephropathy in adults-treatment standard. Nephrol Dial Transplant. 2023;38(11):2464-2473.
- Gutiérrez E, Carvaca-Fontán F, Luzardo L, et al. A personalised update on IGA nephropathy: A new vision and new future challenges. The Nephron Journals. 2020;144(11):555-71.
- Kidney disease: Improving global outcomes (KDIGO) Glomerular Diseases Work Group. KDIGO 2021 Clinical practice guideline for the management of glomerular diseases. Kidney International. 2021;100(4S):S1–276.
- Caravaca-Fontán F, Gutiérrez E, Sevillano ÁM, et al. Targeting complement in IgA nephropathy. Clin Kidney J. 2023;16(Suppl 2):ii28-ii39.
- Jennette JC, Olson J, Schwartz M, et al. Heptinstall’s pathology of the kidney: IgA nephropathy and Henoch-Schonlein purpura nephritis. 6th ed. Lippincott Williams and Wilkins; 2007.
- Uffing A, Pérez-Saéz MJ, Jouve T, et al. Recurrence of IgA nephropathy after kidney transplantation in adults. Clin J Am Soc Nephrol. 2021;16(8):1247-1255.
- Donadio JV, Grande JP. IgA nephropathy. N Engl J Med. 2002;347(10):738-748.










Leave a Reply
You must be logged in to post a comment.