






Reference: March 2025 | Issue 3 | Vol 11 | Page 61
The Centre for Cardiac Risk in Younger Persons in Tallaght University Hospital (TUH), Dublin, (CRYP Centre, also known as ‘The CRY Centre’) is a dedicated service set up to deliver multidisciplinary expertise in the diagnosis and management of inherited cardiac conditions (ICCs), and despite the title, is available to any relevant individual or family aged 16 years or over, with no upward limit.
The service was initially established after a heightened focus on sudden cardiac death (SCD) in otherwise very fit and healthy young people in Ireland following the sudden deaths of Cormac McAnallen and John McCall within four weeks of each other in 2004. It is estimated that, on average, two young people in Ireland under 35 years die every week from SCD, and at least 50 per cent of these deaths may occur due to an underlying genetic cardiac condition. Relatives of these individuals, therefore, may have a 50 per cent chance of inheriting this cardiac condition, and potentially be at risk.
In 2006, the taskforce on SCD that had been established emphasised the need for better emergency responses in the event of such cardiac arrests, as well as better services for identification and treatment of at-risk individuals to reduce the occurrence of cardiac arrest in the first place.
The centre was set up as one of two national referral centres for patients with inherited cardiac pathologies, and plays a role in investigation, management, and appropriate cascade screening for patients and their families. Since the service’s establishment in 2007, the landscape of this field has shifted, with a deeper knowledge of both the mechanisms of these conditions, as well as their overall prevalence within the population.
The CRYP Centre
The CRYP Centre receives referrals from across Ireland, with the majority coming from general practice settings. These are typically individuals with a family history of a first degree relative who has passed away due to SCD, or is living with a potentially inheritable cardiac condition.
We also receive a large number of referrals from our fellow cardiologists regarding clinically affected patients who have been diagnosed with potentially inheritable conditions, and where clarification of the actual diagnosis, genetic testing, and familial cascade screening may prove beneficial.
By the nature of the work we do in exploring patients’ family histories in-depth, we often identify other individuals for cascade screening at the time of our assessments, and supply our patients with an information letter to disseminate among first degree relatives for them to produce to their GP to avail of screening with the centre.
The spectrum of conditions we encounter covers all aspects of ICC, including both cardiomyopathies (most commonly hypertrophic cardiomyopathy (HCM or HOCM), but increasingly, genetic dilated cardiomyopathy (DCM), and occasionally arrhythmogenic cardiomyopathy (ARVC)) and ‘channelopathies’ (‘electrical’ cardiac conditions broadly caused by defective function of ion channels in the cardiomyocyte, most commonly long QT syndrome, but also Brugada syndrome and other rare conditions like catecholaminergic polymorphic ventricular tachycardia (CPVT)).
The versatile structure of our clinics must therefore tailor to a variety of pathologies with nuanced management informed by evidence-based investigation and treatment. Additionally, the sensitive nature and psychological impact of these diagnoses, particularly for those families that have experienced the loss of loved ones to their condition, necessitate that we approach our patient cohort in a manner respectful of their individual experience of their diagnosis.
Review and assessment
In our initial review with a patient, we will explore their immediate and extended family history. In fact, and where possible, we try to collect much of this information in advance of the appointment so that we can tailor investigations to the most relevant concerns. This may reveal individuals with an already confirmed clinical or genetic diagnosis, but often, details may be less specific. If these individuals are happy to share their clinical data with us, and it is pertinent to the investigation and management of their relative, we offer a contact email for them to get in touch.
When a family history includes a sudden unexplained or suspected cardiac death, we will seek to contact the relevant coroner to request the results of their post-mortem examination. In certain circumstances, if the cause of death is deemed to have been cardiac in nature, further correspondence with the relevant pathology department may reveal if there was tissue stored at the time of post-mortem examination that may be considered for posthumous genetic analysis with their next of kin’s consent.
If specific tissue for future genetic testing has not been stored and the death occurred within the last 18 months, we may contact the state laboratory to secure stored toxicology samples which may also be a source of DNA for future genetic testing.
In situations where we are reviewing a family who has lost a loved one to SCD, our investigations are guided by the post-mortem results of the deceased. This would include electrocardiogram (ECG), echocardiogram, and ambulatory rhythm monitoring (Holter) in settings where there appeared to be evidence of a cardiomyopathy at the time of death.
For those whose cause of death has been listed as sudden arrhythmic death (SADS), which by definition is a diagnosis of exclusion – SCD with a structurally normal heart – our differential in organising investigations includes ‘channelopathies’ (such as long QT syndrome and Brugada syndrome), premature conduction disease, and CPVT. These patients will receive a baseline echocardiogram, but their investigations focus more so on abnormal repolarisation and/or arrhythmia, and so include ECG, exercise stress testing (EST) and Holter monitors.
While the role of EST in screening for coronary disease has been discredited, it remains an important test in screening for the above primary arrhythmia syndromes. In further testing of these patients we may employ drug provocation testing with ajmaline and adrenaline to improve sensitivity for diagnosis of Brugada syndrome and CPVT, respectively.
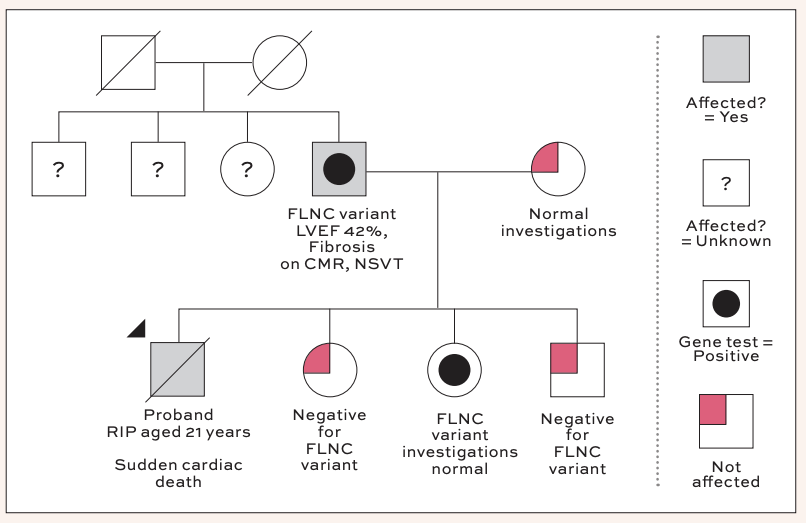
Case example
In discussion with his parents in clinic, it was noted their son had been very well on the night he died. He had been a fit and healthy footballer with no past medical history of note at the time of his passing. He had no recent illnesses or complaints, and in fact, had been out for a run earlier that evening. Clarifying with his parents, it was noted that the deceased was quite tall, and indeed his heart weight may therefore have been normal when indexed to his body surface area.
Clinical testing in the family members was unremarkable, with the exception of the deceased’s father, whose echocardiogram showed mildly reduced left ventricular systolic function. He was then referred for cardiac MRI to assess for the presence of fibrosis. In the interim period, his Holter monitor was returned, demonstrating several short runs of non-sustained ventricular tachycardia (NSVT), with the longest lasting seven beats at a rate of 183bpm.
His subsequent MRI demonstrated a left ventricular ejection fraction of 42 per cent, with a ring-like distribution of sub-epicardial and mid-wall late gadolinium enhancement. At this stage, genetic testing was offered to the patient, and this returned positive for a truncating-variant of FLNC. In light of his family history, Holter monitor, MRI, and genetic results, his case was discussed at our device MDT in TUH, and he was referred for ICD implantation for primary prophylaxis.
Predictive genetic testing was performed in the patient’s three surviving children, with one testing positive for the truncating variant. Investigations for them were reassuringly normal, and they have been linked into the CRY clinical surveillance clinics for routine follow-up. His other two children tested negative for the variant and could be safely discharged. An offer for referral to the CRY Centre has been made to the patient’s remaining first-degree relatives.
Genetic testing
As alluded to previously, the role of posthumous genetic testing is increasingly explored with families when suitable blood or tissue samples are available. This discussion is approached with the deceased’s next of kin by our consultant. Posthumous genetic testing can be of particular utility when clinical testing in living relatives has not elicited any pathology of note.
However, the tissue used for genetic panelling is a precious and finite resource. Although this process has proven to be an invaluable key for many families in answering the question as to why their loved one died – and in turn providing a predictive screening test for their own clinical risk – the likelihood of obtaining an adequate quality DNA sample is influenced by several factors including the sample type, storage, and age of the sample. In cases where a death has been attributed to ‘SADS’, international research has suggested genetic testing of a suitable post-mortem DNA sample may yield an actionable result in 0-25 per cent of cases.
When dealing with a clinically affected patient, we have a thorough conversation with them on the role of genetic testing. Fundamentally, the utility of genetic testing has five key advantages for patients and their families, as outlined by the European Society of Cardiology 2023 Guidelines for the Management of Cardiomyopathies:
1. Clarification of diagnosis –particularly in cases where there is a broader differential based on clinical manifestations which may include a variety of phenocopies (for example, a reported finding of myocarditis on post-mortem may in fact be a manifestation of a desmoplakin gene variant associated with left ventricular variant ARVC, which refocuses family assessment from a zero risk non-hereditary cause to a 50 per cent risk autosomal dominant cause).
2. Prognostication – outcomes may vary depending on the gene (and at times the specific variant within the gene). Being aware of higher risk gene variants allows for appropriate risk stratification and timely intervention, whether an implantable cardioverter defibrillator (ICD) insertion or advanced heart failure referral for transplant consideration; for example, the identification of a lamin A/C gene variant in an individual with DCM will lower the threshold for both ICD implant and early transplant consideration.
3. Therapy – while many inheritable conditions may not yet have gene-specific therapies, we do recognise that this is an ongoing field of research. Moreover, certain genetic diagnoses may also imply additional precautions that should be considered, eg, an SCN5A variant may be identified in an individual with long QT syndrome type 3, but also carries risks of Brugada Syndrome and premature conduction disease. A considerable list of prescription and over-the-counter medications can inadvertently increase the risk of arrhythmia in these individuals if the gene variant is not identified.
4. Reproductive counselling – a genetic diagnosis can be used to inform parents in family planning. It allows for an assessment of the risk of inheritance and also the management of that risk through pre-implantation genetic testing with in vitro fertilisation. For example, a male who is diagnosed with Fabry disease, an X-linked recessive storage disorder affecting the heart, kidneys, and brain, cannot pass this to any male descendants, but all females will be carriers (and may variably manifest signs of the condition).
5. Predictive testing – for those first-degree relatives who are not clinically affected, but who would like to be aware as to their risk of potentially manifesting the pathology during their lifetime, or indeed their risk of passing the variant to their descendants, predictive testing can be offered when a pathological variant is successfully identified in their family.
However, in considering the use of genetic testing, patients should make the decision from a fully informed position, with appropriate guidance, before they give their consent. Psychological aspects which we explore with our patients during this time are whether they may feel different about themselves in a scenario of receiving a positive genetic result, particularly in the setting of those with mild or no obvious clinical manifestations.
Although physically they will not have changed, we recognise that for some individuals this can pose a clear psychological burden – though many of our patients are already keenly aware of their risk of manifesting a condition due to the nature of our clinic and recurrent clinical screening.
Secondly, many of our patients wrestle with the fear that a genetic label may have implications either on their own, or on their family’s, financial futures – particularly when applying for mortgages, life insurance, health insurance, and loans.
Thankfully, we can reassure our patients that under Irish law genetic status is fully protected from misuse. Specifically, the Irish Disability Act 2005 (Section 42, Subsection 2) ensures that genetic data cannot be processed for any of these purposes. This would mean that while a personal history or family history of a clinical cardiac pathology – independent of genetic status – may be declarable and thus informs these financial applications, the presence of a pathogenic gene variant does not.
Moreover, we highlight to our patients that a genetic variant is not sufficient for a clinical diagnosis, but rather reflects a risk of possibly some day developing one.
Along with the utility and implications of gene testing, we also outline the potential results from a genetic test, particularly gene panelling in a proband case, when an appropriate genetic panel is chosen based on clinical phenotype, but where it is not yet defined what specific gene, if any, may be implicated. It is here that we must discuss the potential limitations of genetic testing.
The likelihood of discovering a pathogenic gene variant to explain an individual’s diagnosis varies with their clinical phenotype. For an illustrative example, HCM is one of the most common inheritable cardiac conditions, with an estimated prevalence of 0.2 per cent in the general population, and classically demonstrating an autosomal dominant inheritance pattern.
However, despite readily available testing with a wide array of pathogenic variants, a monogenic cause is only identified in approximately 30-40 per cent of cases, with a remainder believed to possibly be the result of a number of smaller variations in a series of contributory genes which sequentially increase an individual’s vulnerability to manifesting a phenotype.
With this in mind, a patient may be simultaneously both profoundly affected clinically, but yet genotype negative. This can sometimes prove difficult for patients and families to come to terms with, as the condition itself may still present a pattern of inheritance.
However, data published on this specific subgroup of the HCM population in recent years has shown that so-called ‘gene-elusive HCM’ has a lower diagnostic yield with sequential screening in unaffected relatives – particularly when their baseline maximum left ventricular wall thickness on initial screening is <10mm. This may be of some comfort to those relatives concerned about their likelihood of becoming clinically affected over time.
Another possible result which we counsel our patients on before proceeding with genetic panelling is the possibility of a genetic variant of uncertain significance (VUS). This represents a gene variant for which not enough is known about its pathogenicity to determine whether it is causative for a cardiac condition or if it simply represents a non-pathogenic variant.
Unfortunately, given the uncertain nature of a VUS pathogenicity, particularly that it may represent a benign variant found in healthy members of the general population, they cannot be used for the purpose of predictive testing – as without a definite link to a phenotype, a negative result cannot offer reassurance that an individual is not at risk of developing the pathology in question.
Our knowledge of gene variants and their clinical implications is ever-expanding with more and more individuals undergoing testing worldwide, which means that while new gene variants may arise where little is yet known, their classification is dynamic, and so may become clearer over time.
For those clinically unaffected individuals who are found to be carriers of a pathogenic variant – or indeed in a family where a pathogenic variant has not been identified – we schedule ongoing clinical screening for the relevant pathology in our clinic on a routine basis. This can be arranged sooner should any symptoms arise prior to the scheduled appointment.
In specific settings we may offer additional precautions, as appropriate, such as the avoidance of QTc-prolonging medications in those positive for a variant associated with long QT syndrome, or relevant Brugada precautions in those with a pathogenic SCN5A variant.
Psychological impact of ICCs
In addition to the clinical aspects described, there is a considerable psychological impact of ICCs. This is expected in those affected by sudden death of a close relative, but perhaps less anticipated and acknowledged in those diagnosed with a genetic cardiac condition.
While identification of those at risk can allow us to offer risk mitigation (for example, with ICD implant), there are lifelong consequences such as potentially being unsuitable for planned career options, being recommended to reduce sports activity which has been the main focus of social activities and identity, and challenges with family planning.
The CRY-Ireland charity – whose fundraising activities facilitated the original opening of the centre in 2007, the provision of the original dedicated premises in TUH, and the equipping of the current off-campus site of the service – have been able to develop a suite of psychological support services which patients and families can avail of. This support network, running alongside the ease of access to the multidisciplinary team, most particularly our four excellent clinical nurse specialists, goes some way towards lessening the impact of these life-altering conditions.
A family were referred to the CRY Centre for assessment following the tragic death of their son/brother, aged 21 years. In advance of the initial appointment with the family, the deceased’s post-mortem report was requested. This demonstrated a mildly hypertrophied left ventricle with increased heart weight, and evidence of myocardial fibrosis on histology. No other pathology was identified on post-mortem and a concluding diagnosis of myocarditis was made.
References available on request
