






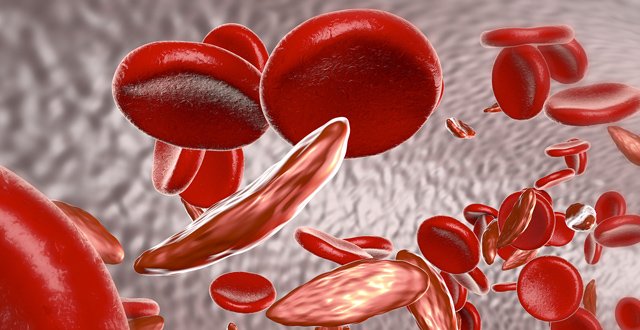
Sickle cell disease (SCD) is an autosomal recessive haemoglobinopathy that we now encounter with increasing frequency in clinical practice in Ireland. The clinical hallmark is a triad of painful vaso-occlusion, micro-infarct end organ damage and a haemolytic anaemia. The disease is characterised by a point mutation in the beta globin gene leading to the substitution of valine for glutamic acid in the beta haemoglobin chain. This seemingly simple amino acid substitution causes a life-threatening and complex multi-organ disease with multiple pathophysiological pathways. Mortality has been high in SCD especially in the early childhood years.
Modern treatment approaches including new-born screening, pneumococcal prophylaxis, transcranial Doppler ultrasound and the introduction of early disease-modifying treatment has seen mortality decrease, the mean age at death increase, and fewer children dying from infection. Aggressive management strategies in childhood yield a sickle-related survival rate of 94 per cent at 18 years and 99 per cent at 16 years. This improvement in childhood mortality has not yet been seen in the adult population who have a median life expectancy in the US of 42 years for males and 48 years for females.
Most patients with SCD will experience vaso-occlusive events at some point in their life and these episodes account for over 90 per cent of all emergency hospital admissions. They can lead to acute organ failure or chronic organ damage affecting all systems. Diagnosis is now typically achieved in the neonatal period due to the introduction of a screening process where haemoglobin electrophoresis is performed on cord blood at delivery. This allows for the early detection of the condition, initiation of parental education and commencement of supportive therapy including antibiotic prophylaxis, pneumococcal vaccination and folic acid supplementation. The morbidity of SCD is highly variable, but can be devastating. Early introduction of disease-modifying treatment is essential to prevent or ameliorate the consequences of this disease.
<h3><strong>Morbidity</strong><strong></strong></h3>
Pain is a commonly seen presentation in children with SCD. The first episode of pain typically affects the small bones of the hands and is referred to as dactylitis. This affects approximately 50 per cent of children with SCD by two years of age. As these children get older the long bones become affected and by adolescence pain is typically associated with the ribs and vertebrae. This pain can be severe and debilitating.
Many other acute complications of SCD have life-threatening consequences. Acute chest syndrome is the most common cause of hospitalisation for patients with SCD, with a peak incidence in early childhood, and is responsible for approximately 25 per cent of sickle-related deaths. The neurological system can also be affected with the occurrence of stroke. Prior to the findings of the STOP study, 10 per cent of children with SCD had a stroke before the end of the second decade of life. Now children are screened with transcranial Doppler ultrasound, which has reduced the stroke risk rate to approximately 1 per cent. Acute splenic sequestration is a life-threatening complication that occurs in up to 30 per cent of patients with SCD up to six years of age. It is characterised by rapid enlarging of the spleen followed by circulatory collapse. All organs of the body are at risk of severely debilitating and life-threatening chronic complications. Some of the more commonly encountered complications include avascular necrosis (AVN), pulmonary hypertension and retinopathy. Avascular necrosis occurs with an incidence of 2.5 per 100 patient years and has even been described in children as young as five years old. It often requires treatment with joint replacement.
<h3><strong>Pathogenesis</strong><strong></strong></h3>
The primary defect in the pathogenesis of SCD is that the resulting haemoglobin is less soluble when exposed to deoxygenated conditions and forms polymers within the red blood cell (RBC). These polymers aggregate causing the RBC to enlarge and deform. This provides it with the characteristic sickle shape. Upon re-oxygenation the RBC cell typically resumes its normal biconcave form, however after numerous episodes of sickling, the cells become irreversibly sickled. These sickle shape cells also are more easily dehydrated when deoxygenated and as a result acquire abnormal intracellular signalling pathways, which activate RBC adhesion molecules on the RBC membrane. These adhesion molecules are involved in the adhesion of RBCs, endothelial cells, neutrophils, monocytes and platelets, inducing the activation of multiple pro-inflammatory pathways. The resulting abnormal RBC has a shorter life span and has increased adhesive interactions. The clinical implication is that the RBC haemolyses and there is a slowing of blood flow in the microcirculation leading to painful vaso-occlusion and micro infarct end organ damage.
<h3><strong>Management</strong><strong></strong></h3>
The management of SCD is multidisciplinary and is comprised of strategies to treat acute sickle cell crisis and long-term disease modifying approaches to prevent complications. This long-term approach is divided into supportive measures, disease modifying treatment and curative options.
<h3><strong>Hydroxyurea/(Hydroxycarbamide)</strong><strong></strong></h3>
Hydroxyurea was approved by the US Food and Drug Administration (FDA) in 1998 and the European Medicines Agency in 2007 for the management of patients with SCD although its use in children is still considered to be off-label by the FDA. It has proven successful in the prevention of complications in SCD and reduces mortality. In spite of our two decades of experience of hydroxyurea we still do not have a full understanding of its mechanism of action. Primarily it affects cell division by inhibiting the enzyme ribonucleotide reductase and thus depletes the cell of deoxyribonucleotide triphosphates (dNTPs). This leads to arrest of cell division or cell death.
Hydroxyurea also works by increasing foetal haemoglobin (HbF) production at the expense of normal adult haemoglobin (HbA). It does this by interfering with beta globin gene expression at the beta globin locus. HbF is the predominant haemoglobin in foetal life and early infancy and binds to oxygen with higher affinity than normal adult haemoglobin. This allows adequate oxygen transfer to the foetus during gas exchange in the intervillous space between mother and baby where there is mixing of both oxygenated and deoxygenated maternal blood. By six months of age it becomes almost entirely replaced by adult haemoglobin. Hydroxyurea reverses this natural post-natal switch from HbF to HbA and this results in reduced polymerisation, sickling, adhesion and haemolysis. The microcirculation blood flow improves and vaso-occlusive events are reduced. Hydroxyurea’s ability to increase nitric oxide further improves blood flow in the microcirculation. Nitric oxide is a potent vasodilator that becomes depleted in haemolysis. Free haemoglobin released by haemolysed RBCs scavenges nitric oxide and depletes it. By reducing haemolysis and inhibiting the scavenging effect of free haemoglobin, the vasodilatory effect of nitric oxide is preserved.
Hydroxyurea is not helpful in the acute management of a crisis as it takes weeks to months to become effective. There is no specific marker in clinical use to determine if the treatment will be of any benefit to the patient and in clinical practice it is offered to any patient who may achieve benefit. Many reproducible studies demonstrate that hydroxyurea reduces mortality with long-term use and reduces long-term complications in both adults and children. The 2011 Baby HUG trial showed that infants treated with hydroxyurea had improved splenic function, reduced pain episodes, reduced chest syndromes, reduced dactylitis and reduced need for transfusion. The treatment is largely very well tolerated and the predominant side effect is cytopaenias. As a result, these patients require regular full blood counts.
<h3><strong>Blood transfusion</strong><strong></strong></h3>
Blood transfusion therapy in SCD is of benefit in both the acute management of vaso-occlusive events and in the chronic management to prevent micro-infarct end organ damage. Most patients with SCD will undergo numerous transfusions in their lifetime putting them at risk for transfusion-associated complications. A simple top-up red RBC transfusion dilutes the volume of sickle cells containing haemoglobin S and replaces them with normal RBCs containing haemoglobin A. The resulting normal RBCs have a longer lifespan than their sickled counterparts. There is feedback to the kidneys to reduce erythropoietin production and the production of new sickle cells is slowed. The transfused blood increases the oxygen saturation of the circulation, increasing oxygen delivery to the tissues. This principle of increasing oxygenation is used in the pre-operative setting where a top-up RBC transfusion to a haemoglobin value of 10g/dL is associated with a reduced incidence of post-operative vaso-occlusive events.
In the long-term management of SCD, reducing the haemoglobin S percentage to below 30 per cent leads to a reduction in vaso-occlusive pain and acute chest syndromes. Many patients with SCD are commenced on chronic transfusion programmes scheduled at patient specific intervals guided by the patient’s clinical findings, haemoglobin level and haemoglobin S percentage. Regular transfusions are required for secondary prevention of stroke, acute chest syndrome, painful events and priapism. The SIT trial randomly assigned children with SCD and silent cerebral infarcts to monthly transfusions versus observation for three years and found that there were significant improvements in the quality-of-life of patients receiving regular transfusions. There were also reduced rates of pain, acute chest syndrome and symptomatic avascular necrosis.
<h3><strong>L-glutamine</strong><strong></strong></h3>
In July 2017 the US FDA-approved L-glutamine oral powder for patients five years and older to reduce the complications of SCD. This was the first drug licensed for the management of SCD since hydroxyurea in 1998. Many studies have demonstrated that RBCs that are sickled have increased susceptibility to oxidative stress. Oxidative stress is measured by changes in nicotinamide adenine dinucleotide (NAD) homeostasis and is defined as the NAD redox potential. NAD redox potential is 20-30 per cent lower in sickle RBCs, but NAD is found in higher levels in sickle cells than in controls. This suggests that in the event of oxidative stress there is increased uptake of NAD by the sickle RBC. Sickle RBCs absorb glutamine, a precursor of NAD at increased rates compared to non-sickle cells. A hypothesis was formed that with increased glutamine supplementation, increased transport and utilisation of glutamine in sickled RBCs will lead to increased NAD and NADH levels thus providing increased defence against oxidative stress. The benefits of administering both L-glutamine and hydroxyurea together appear to be greater than either agent alone.
<h3><strong>Crizanlizumab</strong><strong></strong></h3>
It is known that activation of the inflammatory system plays an important role in the pathogenesis of SCD. Cytokines are released and they quickly activate white blood cells. These activated white and red blood cells then bind to the endothelial cells via several selectins and integrins expressed by endothelial cells. Adhesion of inflammatory and red blood cells to the endothelium is primarily initiated by P selectin. Both animal and human studies have identified that deficiency of P selectin in those with SCD leads to defective adhesion of white blood cells to endothelial walls. This ultimately protects from vaso-occlusion. There are numerous potential treatments targeting P selectin in various stages of investigation.
One of promise is crizanlizumab. It is a humanised monoclonal antibody that binds selectively to P selectin. A phase 2 multicentre randomised controlled trial known as SUSTAIN was reported in the <em>New England Journal of Medicine</em> (<em>NEJM</em>) in 2016. In this trial high-dose treatment was associated with reduced frequency of sickle cell pain events compared to placebo. The annual sickle cell-related pain event rate was reduced by 32.1 per cent in patients already treated with hydroxyurea and by 50 per cent in those who were not already receiving hydroxyurea. The treatment was well-tolerated and there is much promise for this therapy as it undergoes further investigation
<h3><strong>Haematopoietic stem cell transplant</strong><strong></strong></h3>
Haematopoietic stem cell transplant (HSCT) is recognised as a potentially curative treatment for SCD. The current gold standard is myeloablative HLA matched sibling donors due to the reduced risk of both graft versus host disease and graft rejection. There have also been approaches using less myeloablative conditioning regimens. There are trials ongoing exploring the use of unrelated donors and despite the successes of this technique in the treatment of malignant blood disorders, results in SCD patients have largely had unsatisfactory results. The major difficulty in employing HSCT as a curative treatment for patients with SCD remains patient selection. This is attributed to disease heterogeneity, challenges in identifying patients at high risk of developing advanced disease, patient eligibility due to advanced multi-organ disease and concerns relating to transplant-related mortality. In clinical practice the experience of HSCT and SCD has yielded a five-year survival rate of 90-97 per cent, a transplant-related mortality rate of 7-10 per cent and an SCD-free survival rate of 80-90 per cent. The role of early transplantation in children to reduce SCD mortality has not been fully explored, but remains an area of ongoing investigation. HLA-haploidentical transplant is an appealing treatment due to the expansion of the donor. A ‘half matched’ related donor, usually the mother or father is used. An optimal conditioning regimen has not yet been identified, but it remains an area of advancing research.
<h3><strong>Gene therapy</strong><strong></strong></h3>
Less than 18 per cent of eligible patients have a HLA-matched sibling donor for transplant. As a result, gene therapy is an increasingly exciting area of research in SCD and it may provide a long-term, potentially curative, treatment. Successful, sustained gene therapy has been reported previously in mouse models by lentiviral transfer of an anti-sickling beta-globin variant. In 2017 the <em>NEJM</em> published the outcome of a 13-year-old boy who was enrolled in a clinical trial to receive gene therapy using a LentiGlobin BB305 vector. This lentiviral vector is currently being used in seven patients with SCD and 22 patients with beta thalassaemia. It has a stable safety profile and has no evidence of insertional oncogenesis at 30 months. In this boy the transduced cells engrafted and normal cell counts followed across all lineages by day 88 post-transplant. The investigators did not report any vector-related side effects and the child remains well.
<h3><strong>Summary</strong><strong></strong></h3>
The complications of SCD are devastating to patients and until now progress has been slow in the achievement of cure. Thankfully, due to the recent advances in our knowledge of SCD pathogenesis we have, however, reached an exciting time. We have more potential treatment targets than ever and it is likely that considering the numerous pathological mechanisms involved, optimal therapy will require a multi-targeted treatment approach. Dynamic international collaboration and availability of funding is essential to support the ongoing research required to treat this highly complex disease.
<strong>References on request</strong>










Leave a Reply
You must be logged in to post a comment.